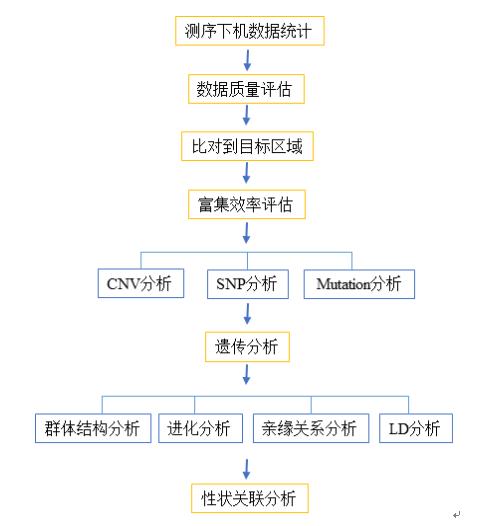
|
目标区域测序技术介绍
***************************************************************************************************** 技术简介: 目标区域测序(Targeted Sequencing):是指针对感兴趣的目标区域富集后进行大规模测序。研究者可以针对自己感兴趣的染色体区域或者大量的候选基因区域进行数百个甚至上千个样品的序列测定。 技术优势
应用领域
技术参数与实验流程
***************************************************************************************************** 技术参数
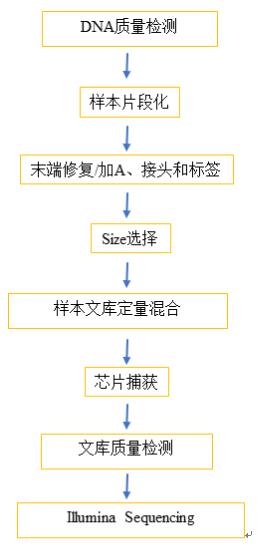
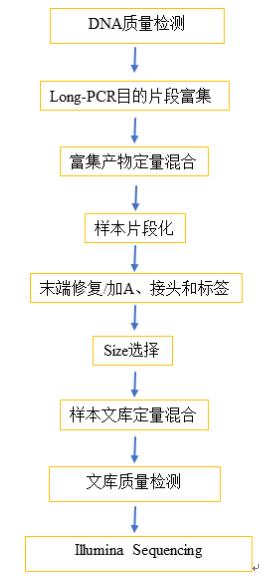
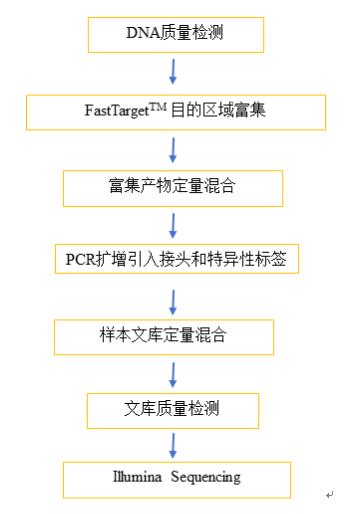
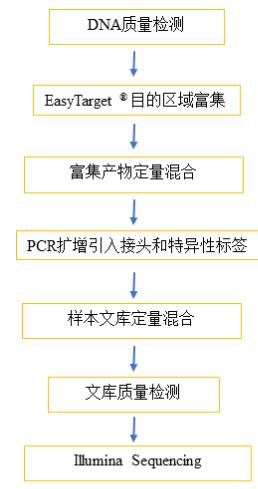
实验流程
经典案例解读
***************************************************************************************************** 案例1:基于目标区域测序策略进行奶牛产奶性状功能基因挖掘 背景: 全基因组关联分析(GWAS)己经广泛地应用于QTL性状的研究当中,并且定位出了大量显著的SNP位点,然而这些标记位点并非真正影响目标性状的功能变异,而可能与功能变异处于连锁状态,因此可以通过对 GWAS 鉴定的区域进行目标区域测序,从而找到与QTL性状紧密相关的功能变异。 方法: 研究人员前期对中国荷斯坦牛群体进行了产奶性状的GWAS研究,定位到105个显著SNP位点,选取6.6Mb的显著区段,运用目标区域捕获测序(安捷伦Sure select)对60头公牛进行高通量混池测序(随机每6头牛一个文库), 测序深度为131×。 结果: 通过目标区域测序鉴定到127218个SNP,其中735位于编码区,418位于UTR区。 经过分析和筛选,选取了200个SNP在来自北京的17个公牛家系和上海的15个公牛家系共734头母牛上运用质谱技术进行基因分型。 通过对产奶性状进行关联分析,发现分布在53个基因上的66个SNP与奶牛产奶性状显著关联,在这53个基因中,其中26个基因的SNP与前期GWAS结果一致。 进一步选择了其中20个显着基因来分析它们在不同组织中的基因表达水平,发现15个基因在乳腺中是特异高表达的。 参考文献:Li Jiang., et al., Targeted resequencing of GWAS loci reveals novel genetic variants for milk production traits. BMC Genomics, 2014 Dec 15;15:1105. 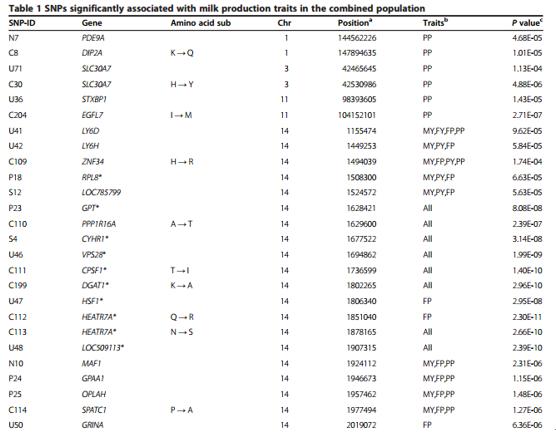
表一:与奶牛产奶性状显著关联的SNP。a: 基于奶牛UMD_3.1 基因组序列. b: MY = 产奶量, FY = 脂肪产量, PY = 蛋白产量, FP =脂肪百分比, PP =蛋白质百分比。 *: 这些基因用于基因表达量检测。
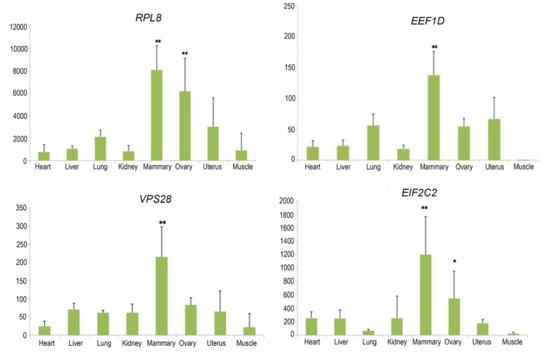
图二:4个泌乳期奶牛的八个组织中基因的相对mRNA表达水平。
案例2:基于目标区域测序策略进行物种分类和系统进化分析 背景:不同鸟类的演化仍然有争议,需要进行更好的物种分类。 方法:研究人员利用杂交富集的定向测序技术,对198种现存鸟类(代表所有鸟类谱系和两个鳄鱼外群)的394保守位点进行目标区域测序,然后基于测序数据使用贝叶斯法和最大似然分析法建立所有鸟类谱系的系统进化树。 结果: 产生259个高质量测序核位点(平均长度为1523个碱基)共7.8 × 107 个碱基的数据量。 使用贝叶斯法和最大似然分析法建立所有鸟类谱系的进化树高度一致,5个主要分支形成新鸟纲 (Neoave) 的连续姐妹类群:(1)包括夜鹰,雨燕和蜂鸟;(2)包括杜鹃,大鸨,鸽子,蕉鹃和沙鸡;(3) 鹤及其亲属;(4)水鸟类群,包括潜水类、涉水类、岸滩类;(5)麝雉类。 进化分歧时间与古生物记录一致,但是不支持最近提出的 “新鸟纲”两个分支Columbea和Passerea。 参考文献:Li Jiang., et al., Targeted resequencing of GWAS loci reveals novel genetic variants for milk production traits. BMC Genomics, 2014 Dec 15;15:1105. 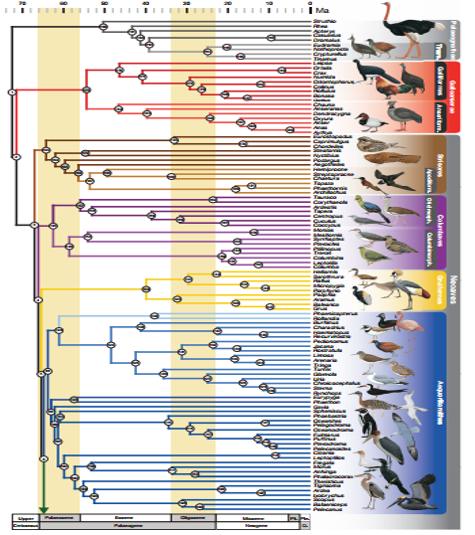
图一:鸟类的系统发育树
|